Übersetzung von Auszügen eines 2009 erschienenen Artikels über das Rätsel des zusätzlichen X-Chromosoms:
[…]
Kreislauf der Gonadotropine
FSH und LH werden durch die Hypophyse freigesetzt und wandern beide durch den Blutstrom zu den Hoden, wo FSH die Spermienproduktion und LH die Testosteronproduktion anregt. Bei normalen Männern signalisiert das erzeugte Testosteron im Blutstrom (gemeinsam mit einem weiteren Protein namens Inhibin) der Hypophyse, dass FSH und LH richtig arbeiten. Als Reaktion verringert die Hypophyse die Ausscheidung von FSH und LH, bis die Testosteronwerte unter einen kritischen Punkt sinken. Diese Feedbackschleife ist ein übliches Phänomen der endokrinen Selbstregulierung: Bei normalen Männern ist es die Art der Hoden, der Hypophyse zu signalisieren, dass die Botschaft angekommen ist und sie ihren Job erledigen. Inneres Feedback hält die Testosteronwerte im Kreislauf in einem eng begrenzten Normalbereich – ideal für die sexuelle Funktion und den körperlichen Stoffwechsel.
Beim Klinefelter-Syndrom werden FSH und LH normal erzeugt, aber als Reaktion wird Testosteron ungenügend erzeugt, um normale Hodenfunktionen zu gewährleisten und der Hypophyse eine negative Rückmeldung zu geben. Die Hypophyse erkennt einen niedrigen oder nicht vorhandenen Testosterongehalt im Blut und bombardiert die Hoden im Gegenzug mit FSH und LH – als ein Versuch, die Testostonerwerte anzuheben, aber erfolglos.
Die Samenkanäle der Hoden – wo Spermien und Testosteron erzeugt werden – sind atrophiert und besitzen keine Funktion bei Männern mit Klinefelter-Syndrom. Die Hypophyse, die – weit von den Hoden entfernt – vom Gehirnstamm baumelt und die vergebenen Mühen ignoriert, produziert weiterhin große Mengen FSH und LH. Deshalb zeigt die klassische Hormonprobe eines erwachsenen Klinefelter-Patienten deutlich erhöhte FSH- und LH-Werte bei verschwindend geringem Testosteron.
Das Rätsel der X-Inaktivierung
Dem ersten Bericht über einen Klinefelter-Patienten im Jahr 1942 ( von Dr. Klinefelter und Kollegen) folgte 17 Jahre später die Entdeckung der genetischen Herkunft des Syndroms. Im normalen menschlichen Genotyp enthält jede Zelle 46 Chromosomen, inklusive beider Geschlechtschromosomen: Entweder XX oder XY. Deshalb ist der normale weibliche Genotyp 46,XX, während der normale männliche 46,XY ist. Die klassische genetische Anomalie bei Klinefelter ist ein zusätzliches X-Chromosom, welche als 47,XXY-Genotyp beschrieben ist (weitere Formen existieren, sind aber viel seltener)
Das zusätzliche Chormosom erscheint als Ergebnis der nicht vorhandenen Trennung während der Meiose: Ein zusätzliches X-Chromosom schleicht sich herein, wo es während dem Prozess der Zellteilung nicht hingehört, wenn väterlicher Samen oder mütterliche Eizellen erzeugt werden. Aufgrund der Verbindung von Samen und Eizelle entsteht ein XXY-Embryo.
Unter genetischen Gesichtspunkten bestimmt das vorhandene Y-Chromosom das männliche Geschlecht. Das Y-Chromosom enthält eine Region namens SRY (sex determining region), welches für die molekularen Signale verschlüsselt, die die Entwicklung der männlichen Keimdrüsen und nachfolgender Testosteronproduktion einleitet, sowie die Entwicklung der innen- und außenliegenden Genitalien. Nachdem sie ein Y-Chromosom besitzen, sind Individuen mit Klinefelter-Syndrom im Genotyp eindeutig männlich. Aber phänotypisch sind die klassischen Erscheinungsformen des männlichen Geschlechts beim Klinefelter-Syndrom verschoben, offenbar durch das vorhandene, zusätzliche X-Chromosom aus dem Gleichgewicht gebracht.
Die Rolle des zusätzlichen X-Chromosoms ist jedoch komplizierter als angenommen:
Das Rätsel für Ärzte und Forscher liegt in einer Feinheit dessen, wie sich Geschlechtschromosomen äußern, X-Inaktivierung genannt. Bei normalen Zellen, die mehr als ein X-Chromosom enthalten (wie bei normalen weiblichen XX-Zellen), ist ein X immer inaktiv. Diese Inaktivierung geschieht frühzeitig bei der embryonalen Entwicklung und hinterlässt nur ein funktioniertes X in jeder Körperzelle. Das verbleibende, inaktivierte X wird „Barr-Körperchen“ genannt, und seine Gene tragen nicht zur Funktion dieser Zelle noch zur Funktion des Menschen bei.
X-Inaktivierung ist dafür bekannt, dass sie in allen Zellen stattfindet, die mehr als ein X-Chromosom enthalten. Das ist für Klinefelter-Forscher bedeutsam, denn es ist ebenso auf die Zellen der XXY-Männer anwendbar. Die Frage bleibt: Was unterscheidet den tatsächlichen Genotyp eines Mannes mit Klinefelter-Syndrom von dem eines normalen Mannes? Nach der X-Inaktivierung in einer XXY-Zelle verbleibt die Anzahl der aktiven Gene bei 46, einschließlich einem X, einem Y und einem inaktiven X – dem Barr-Körperchen.
Eine Teilantwort liegt darin, was über das Barr-Körperchen in normalen weiblichen XX-Zellen bekannt ist: Obwohl das Barr-Körperchen inaktiv genannt wird, gilt die Inaktivierung des zweiten X-Chromosoms nicht vollständig. Eine kleine Zahl seiner Gene bleibt wirksam. Bei XXY-Männer verursacht eines oder mehrerer dieser verbleibenden, aktiven Gene zweifellos die weiblichen, sekundären Geschlechtsmerkmale, atrophierte Kanäle und resultierende Unfruchtbarkeit, aber der exakte molekulare Mechanismus bleibt unbekannt.
[…]
Ein Zitat einer Mutter eines Kindes mit Klinefelter-Syndrom weiter unten im Artikel:
Sie nennt es einfach „XXY“ – „Ich hasse das Wort ‚Syndrom‘ – es klingt danach, als stimmt mit ihm etwas nicht.“
***
Ab wann kommt der Testosteronmangel zum Tragen?
Viele Fachärzte und auch Betroffene in der Klinefelter-Community sind der Meinung, dass auffälliges Verhalten von XXY-Männern ausschließlich auf den Testosteronmangel zurückführbar ist.
Die Medizinische Universität von South Carolina führt folgende Grenzbereiche für Testosteron für 46,XY-Männer an:
Erwachsener (Früh): 9.36-37.1 nmol/L
Frühgeborene: 1.28-6.87 nmol/L
Neugeborene: 2.6-13.9 nmol/L
Kinder, 1 to 5 Monate: 0.03-6.14 nmol/L
Kinder, 6-11 Monate: 0.07-0.24 nmol/L
Kinder, 1 to 5 Jahre: 0.07-0.87 nmol/L
Kinder, 6 to 9 Jahre: 0.10-1.04 nmol/LTanner-Stadien:
I: 0.07-0.8 nmol/L
II: 0.17-2.43 nmol/L
III: 0.52-9.72 nmol/L
IV: 3.64-18.91 nmol/L
V: 9.19-27.76 nmol/L
Die Werte stammen dabei von Studien aus dem Jahr 1995 (Kinder und Heranwachsende) bzw. von 2004 (Erwachsene).
Die einzelnen Tanner-Stadien beziehen sich dabei auf die Pubertätsphasen. Insgesamt lässt sich feststellen, dass die Testostonwerte im Kindesalter verschwindend gering sind, und erst im Laufe des III. Stadiums (entspricht ca. 10-14 Jahre) deutlich zunehmen.
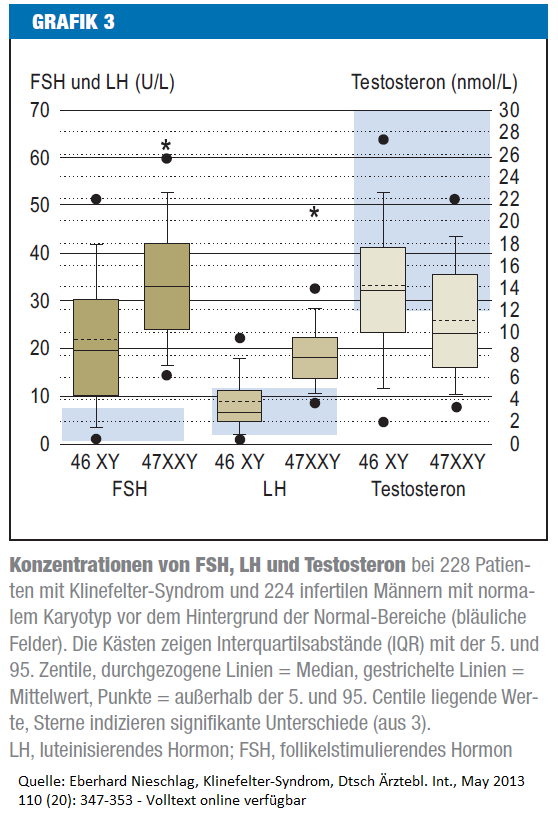
Die Abbildung zeigt die Durchschnittswerte von 228 Männer mit Klinefelter-Syndrom und 224 unfruchtbaren Männern mit normalem Karyotyp, blau wäre der Normalbereich (12-30 nmol/L)
Die XXY-Männer bewegen sich zwischen 7 und 15 nmol/L, also teilweise im Normbereich, wobei der Median bei 10 nmol/L liegt. (Quelle: Nieschlag 2013)
Im Vergleich zur obigen Tabelle aus den USA fällt auf, dass der untere Schwellenwert für XY-Erwachsene seit 2004 um 3 nmol/L angehoben wurde. Ob es dafür eine tatsächliche größere medizinische Bandbreite als Ursache gibt oder ob die Pharmakonzerne von der Anhebung der Grenzwerte profitieren, weil dann rascher substitutiert wird, kann ich nicht beurteilen. Der Normbereich ist jedenfalls sehr variabel und wann er von XXY-Männern verlassen wird, ist individuell sehr verschieden.
Vorläufiges Fazit ist jedenfalls, dass sich der Testosteronmangel bei XXY-Männern offenbar frühestens ab dem ca. 10. Lebensjahr bemerkbar macht, je nach Abfall des Testosteronspiegels rutscht er auch erst nach der Pubertät bzw. im jungen Erwachsenenalter unter den Normbereich.
*
Meine Schlussfolgerung als Laie:
Ich kann mir nicht erklären, wie Testosteronmangel bereits im Vorschulalter Auffälligkeiten wie Schüchternheit, Passivität, motorische Defizite und Verzögerung bei der Sprachentwicklung erklären soll, wenn zu diesem Zeitpunkt aufgrund der verschwindend geringen Konzentrationen an Testosteron noch gar kein Mangel besteht bzw. offenbar vernachlässigbar ist.
Bis zu diesem Zeitpunkt würde ich laienhaft vermuten, dass die Ursache im zusätzlichen X-Chromosom bzw. in dessen verbleibenden, aktiven Genen zu suchen ist. Und je nachdem, wie viele Gene noch aktiv sind, reicht die Ausprägung dann hin zu Entwicklungsstörungen wie ADHS, Autismus, Schizophrenie, usw…. , oder es wurden so viele Gene inaktiviert, dass kaum Symptome vorhanden sind. Hinzu kommen natürlich noch erbliche Faktoren und erlerntes Verhalten durch die Eltern.
Was bisher auch durch mir bekannte Studien nicht widerlegt wurde:
Das schlechte Kurzzeitgedächtnis, beeinträchtigte Exekutivfunktionen (präfrontaler Cortex) sowie die sensorische Überempfindlichkeit bleiben auch nach der Substitution oft erhalten – Symptome, die sowohl auf ADHS als auch auf Autismus hindeuten, also im Gehirn ähnliche Bereiche betreffen könnten. Ebenso hat die Substitution keinen Einfluss auf die verstärkte Detailwahrnehmung allgemein, auf visuelles Denken und Langzeitgedächtnis.
Positive Effekte sind dagegen bei der Konzentrationsfähigkeit sowie beim Sprachfluss beobachtet wurden, was etwas im Widerspruch zur oben genannten verzögerten Sprachentwicklung im Kindesalter steht. Wobei mir auch nicht klar ist, ob die Schüchternheit Folge der generellen Mutlosigkeit ist, oder der beeinträchtigten Exekutivfunktionen (Gedankenspiralen, wie sagt man etwas, in welcher Reihenfolge, wie gehe ich auf jemanden zu, etc….), ich möchte das zumindest in Frage stellen, dass das schüchterne Erscheinungsbild bloß die Folge eines Hormonmangels ist.
Vielleicht finde ich auf dieses Rätsel noch Antworten in den zahlreichen Artikeln, die ich gefunden, aber noch nicht durchblickt habe. Würde mich ein Spezialist daraufhin weisen, wo mein Denkfehler liegt, ginge es natürlich schneller. Ob Bekräftigung oder Ablehnung meiner Theorie, spielt keine Rolle, ich möchte nur Beweise sehen, und kein pauschales „So ein Blödsinn kann nur von einem nichtstudierten Laien kommen“.
