Auf diesem Blog ist es ruhig geworden, vor allem mangels neuen Erkenntnissen, aber auch der deprimierten Feststellung, dass sich am geringen Wissensstand über Klinefelter seitens der Ärzte wenig ändern wird. In Österreich gilt das noch mehr als in Deutschland, weil Österreich ein medizinisches Entwicklungsland ist. Sich seinen (neuen) Ärzten bezüglich neuen Erkenntnissen über Klinefelter und zusätzlicher Diagnosen mitzuteilen, ist auf mündlichem Weg fast nicht möglich. Die Katze beißt sich hier natürlich in den Schwanz, denn mit einer kommunikativen Störung fällt es mir noch schwerer, in wenigen Worten auf den Punkt zu kommen. Ich müsste zu weit ausholen und kein Arzt nimmt sich soviel Zeit für mich. Lediglich auf schriftlichem Weg kann ich darauf hinweisen, wobei ich dabei Gefahr laufe, zu ausschweifend zu schreiben.
Eine weitere Frage, die ich sehr früh hier thematisiert habe, ist, ob man sich seinem Arbeitgeber gegenüber outen soll. Anfangs habe ich das neutral gesehen, inzwischen glaube ich, dass die Vorgesetzten von so einer Diagnose heillos überfordert und unnötige Ängste und Vorurteile geschürt werden. Für mich persönlich haben die -als typisch geltenden – Auswirkungen des Klinefelter-Syndroms und auch die damit verbundene Therapie, die Testosteronzufuhr, keinen direkten Einfluss auf meine Leistungsfähigkeit. Wie viele X-Chromosomen ich besitze, merkt man mir in der Arbeit nicht an. Ob ich Kinder kriegen kann, und warum ich so dünne Arme habe, spielt keine Rolle, allenfalls ist man wiederkehrend mit den schmerzhaften Aussagen kommentiert „Auch Du wirst irgendwann Familie haben.“ oder „Warte mal, bis Du Kinder hast.“, etc… Man weiß genau, dass das nie der Fall sein wird, außer man findet eine Partnerin mit Kindern. Über das Klinefelter-Syndrom ist öffentlich NICHTS bekannt. Wiederkehrende Artikel in diversen Print- und Online-Medien fokussieren rein auf die Unfruchtbarkeit und auf den Mangel an männlichen Merkmalen – nichts, worüber man gerne mit Kollegen und Vorgesetzten sprechen oder gar als Einstieg für eine Offenlegung benutzen möchte. Die Hintergründe dieses Syndroms sind so komplex, dass es wissenschaftlich noch nicht komplett erforscht, geschweige denn verstanden wurde. Wie soll man etwas erklären, was nicht erklärbar ist?
Als ich hier anfing zu bloggen, stieß ich zuvor erstmals auf einen für mich bahnbrechenden Artikel über einen Zusammenhang zwischen Klinefelter und Autismus. Ich passte nicht ins Schema F vieler Klinefelter-Betroffene und konnte mich mit diesen nicht identifizieren. Mit Autismus bzw. dem Asperger-Syndrom konnte ich es. Inzwischen, drei Jahre später, kenne ich *einige* Fälle, wo beides auftritt. Heute stieß ich auf einen Zeitungsartikel in der seriösen britischen Zeitung „The Guardian“ (abgerufen am 10.7.2017):
„Felix has a genetic disorder, Klinefelter syndrome, and is on the autism spectrum, meaning he struggles to deal with sensory overload. The stench – along with an unwelcome night-time accompaniment of jackhammers and concrete saws – sends him into meltdown.
“The constant noise, the constant smell – it actually is having such an impact,” Waters said. “He will literally throw himself on the floor and have a tantrum because to him this is an overwhelming sensory impact. He doesn’t have the cognitive ability to say this will go away in a day or so. He’s trapped in his own mind.”
Der Betroffene hat das Klinefelter-Syndrom und ist im autistischen Spektrum, d.h., er hat Probleme im Umgang mit Reizüberflutung. Ständiger Lärm und Gestank bedeuten für ohne einen überwältigenden sensorischen Einfluss, sodass er sich regelrecht auf den Boden wirft und einen Wutanfall bekommt. Er besitzt nicht die kognitive Fähigkeit zu sagen, dass es nach einem Tag oder so weggehen wird. Er ist in seinen eigenen Gedanken gefangen.
Erdrückende Belege für einen genetischen Zusammenhang:
Ich möchte nachfolgend erläutern, weshalb ich die folgenden Abbildungen für so wichtig halte.
1 Autismus-Spektrum-Fragebogen
Das Setting ist einfach: Eine Kontrollgruppe (gemeint sind Menschen mit normaler Chromosomenanzahl) und Klinefelter-Männer wurden mit dem autistischen Fragebogen (AQ nach Baron-Cohen) getestet. Der Schwellenwert, ab dem ein Screening für Autismus in Betracht gezogen werden sollte, liegt bei 26. Die Kontrollgruppe schnitt durchwegs unter 26 ab, die Mehrheit unter 15. Die Klinefelter-Personen hingegen mehrheitlich über 20, meist über 25.
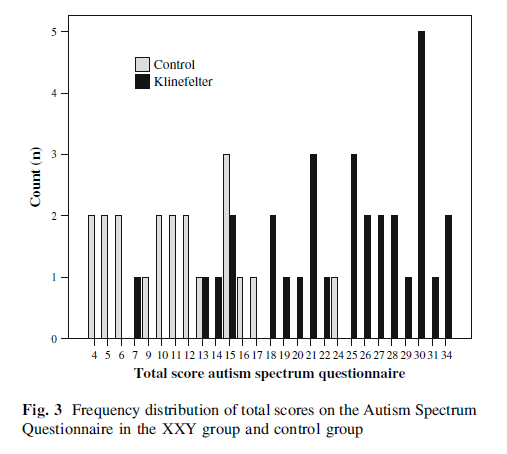
Van Rijn S. et al., Social Behaviour and autistic traits in a sex chromosomal disorder: Klinefelter (47 XXY) syndrome, J Autism Dev Disord, 2008, 38: 1634-1641
2 ADI-R Score: Die Kernbereiche von Autismus
Domain I betrifft soziale Interaktion, Domain II Kommunikation und Domain III stereotype, wiederkehrende Verhaltensmuster/Interessen. Untersucht wurden 51 Klinefelter-Personen. Es hat sich gezeigt, dass Klinefelter-Personen in den beiden Kernbereichen von Autismus, Kommunikation und Interaktion, gleichermaßen betroffen sind. Im Gegensatz zum klassischen Autismus (also keine bekannte genetische Ursache) sind eingeschränkte Verhaltensmuster schwächer betroffen. Das kann bedeuten: Sie zeigen ihren Autismus seltener nach außen, indem sie weniger mit den Händen flattern, zappeln, schaukeln, weniger eng begrenzte Interessensgebiete aufweisen, etc. Für die Diagnose kann das jedoch heißen, dass Klinefelter-Autisten häufiger unerkannt bleiben, weil sie nicht die typischen nach außen hin sichtbaren autistischen Merkmale zeigen.

Quelle: Bruining H. et al., Psychiatric Characteristics in a self-selected sample of boys with Klinefelter Syndrome, Pediatrics, 2009, 123, e865
3 Überschneidungen von Klinefelter mit Autismus
Ein Leitartikel von Lehnhardt et al (2013) geht über typische Autismus-Symptome. Ich habe damals typische Beschreibungen von Klinefelter-Kindern mit denen in der Tabelle verglichen und kam zu dem verblüffenden Ergebnis, dass in allen Punkten eine Übereinstimmung besteht:

Quelle: Lehnhardt et al., Diagnostik und Differential-Diagnose des Asperger-Syndroms im Erwachsenenalter, Dtsch Arztebl Int., 2013, 10 (45): 755-763
Für Erwachsene sieht das anhand zahlreicher Beispiele in der Symptomatik so aus:
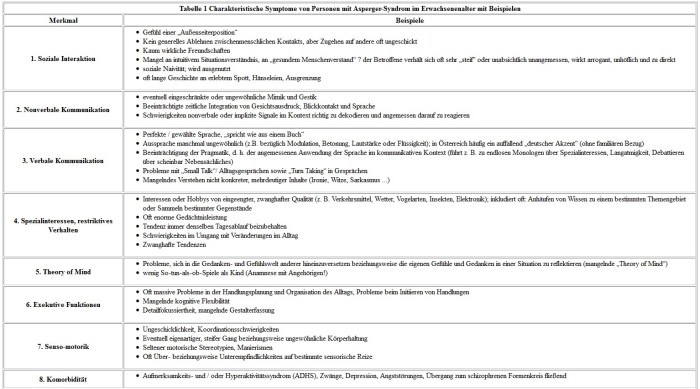
Quelle: http://www.springermedizin.at/artikel/16431-das-asperger-syndrom-bei-erwachsenen
4 Einordnung von Personen mit zusätzlichen X-Chromosomen im autistischen Spektrum
Hier hat man Mädchen und Buben mit zusätzlichem X-Chromosom zusammengefasst, also sowohl 47,XXX als auch 47,XXY. Es gibt zwischen beiden Gruppen einige Übereinstimmungen, was darauf hinweist, dass nicht das Testosteron(defizit) hier den Unterschied macht, sondern das zusätzliche X-Chromosom die entscheidende Rolle spielt. Autismus ist keine Ja/Nein-Frage, sondern eine dimensionale Größe, d.h., sie reicht von vollkommen unautistisch bis schwer autistisch. Der SRS-Score (Social Responsiveness Score) gibt die Anzahl der autistischen Verhaltensweisen an, der Schwellenwert für eine Autismus-Diagnose liegt bei 70:
Autisten ohne zusätzliches X-Chromosom („ASD“) befinden sich mit 95 deutlich darüber.
Die nichtautistische Kontrollgruppe liegt mit 25 weit darunter.
Die XXX/XXY-Gruppe erreicht mit 65 einen Wert knapp unter dem Schwellenwert. Sie befinden sich also im Durchschnitt viel näher an einer Autismus-Diagnose als an einer Nicht-Diagnose.
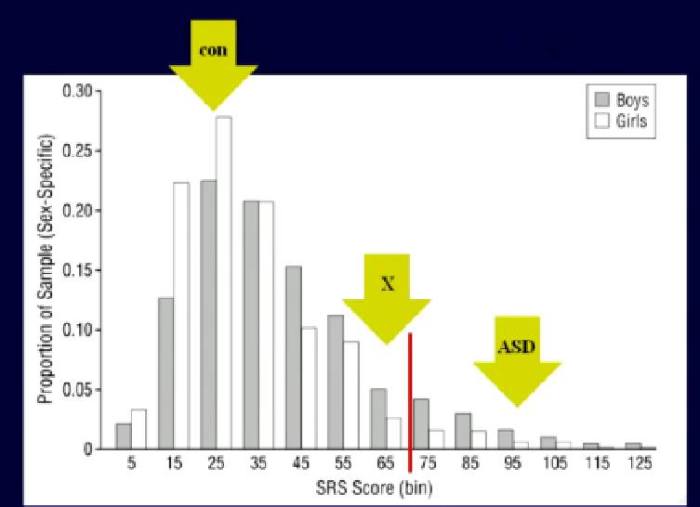
Quelle: Auswirkung des zusätzlichen X-Chromosoms
Ich stelle die These auf, dass Klinefelter lediglich eine von mehreren Auswirkungen des zusätzlichen X-Chromosoms ist. Klinefelter ist nicht 47,XXY und 47,XXY ist nicht Klinefelter. Bei jenen 47,XXY-Betroffenen, die nicht autistisch sind oder so wenig autistische Merkmale aufweisen, dass sie nicht für eine Autismus-Diagnostik in Frage kommen, hängt es von der Aktivität der Gene auf dem zweiten X-Chromosom ab.
Meine Literaturliste zu 47,XXY/Klinefelter-Syndrom ist die wahrscheinlich umfangreichste im deutschsprachigen (und englischsprachigen) Raum. Im Gegensatz zu den Fachleuten bin ich nicht an einen spezifischen Fachbereich gebunden und sehe leichter unerwartete Verbindungen zu den anderen Ursachen und Auswirkungen. Dafür mangelt es mir natürlich an Tiefe in den Fachgebieten selbst und am Verständnis komplexer genetischer und neurologischer Zusammenhänge.
Aufgrunddessen halte ich es in meinem eigenen Fall für zielführender, die Asperger-Thematik anzusprechen, wenn es um Offenlegung von Diagnosen geht. Strenggenommen fußt meine Asperger-Diagnose auf dem zweiten X-Chromosom, aber Ursachen interessieren die wenigsten, die Auswirkungen sind das Thema und da besteht in der Literatur eine Fülle an Material über Asperger, aber nahezu nichts (Verständliches) über Klinefelter.
So, ich möchte meine Leser nicht mit meinen Ausschweifungen über Asperger langweilen, weil das ja nicht alle Auswirkungen bei 47,XXY betrifft.
Darum noch kurz zu den physischen Aspekten dieser Diagnose. Idealerweise finden die hier geschilderten Kontrollen statt. In der Praxis wissen aber sehr viele Ärzte nicht, dass die bloße Zugabe von Testosteron etwa nicht ausreicht, um den Abbau der Knochendichte zu verzögern (aufhalten lässt er sich leider nicht), sondern eine zusätzliche Gabe von Vitamin-D notwendig ist. Ansonsten gilt zur Therapie gegen Knochenschwund das, was man bei vielen Erkrankungen des Bewegungsapparats empfiehlt: Viel Bewegung, genügend trinken, wenig/kein Alkohol und gesund ernähren. Das betrifft gleichermaßen das Risiko aufgrund des gestörten Fettstoffwechsels an Diabetes zu erkranken. Über Chancen und Risiken einer Testosterontherapie, auch im Hinblick auf die Genderidentität und bei intersexuellen Betroffenen (wenige, aber vorhanden), habe ich hier ausführlich geschrieben. Eine leichte Entscheidung, frühzeitig mit Testosteron zu beginnen, ist es nie. Randnotiz: Subjektiv kommt mir vor, als sehe ich heute im Alltag wesentlich mehr Menschen mit – auf den ersten Blick – unklarer Genderidentität, sprich, man weiß nicht, obs ein Manderl oder Weiberl ist, also noch vor zwanzig Jahren. Die Gesellschaft öffnet sich und das Angebot für Betroffene wächst, wie hier in Österreich.
